
Super-Resolution Microscopy
Super-resolution microscopy techniques are so-called because of their ability to resolve structures beyond the diffraction limit of light. Conventional light microscopy techniques are unable to bypass this limit, which prevents structures finer than roughly half the wavelength of the emission light (typically no less than ~200 nm) from being resolved. In contrast, super-resolution techniques have been shown to achieve single nanometer resolution.
Super-resolution imaging reveals more information about biological structures than conventional light microscopy by allowing smaller features to be visualized and in more detail. There are multiple types of super-resolution microscopy techniques, each with their own advantages and disadvantages.
For a quick overview of what super-resolution microscopy is, take a look at our short article: What is Super-Resolution Microscopy.


Super-Resolution Localization Microscopy
One family of super-resolution techniques is the super-resolution localization microscopy techniques, which comprise Photoactivated Localization Microscopy (PALM), Stochastic Optical Resolution Microscopy (STORM) and DNA-based Point Accumulation for Imaging in Nanoscale Topography (DNA-PAINT).
These techniques are able to temporally isolate single fluorophores from a group and take advantage of our ability to localize these fluorophores to sub-diffraction-limited spots. By individually localizing single fluorophores and reconstructing them into one image, a super-resolved image can be generated that effectively breaks the diffraction limit.
SIM and iSIM
Structured Illumination Microscopy (SIM) and the high-speed alternative instant SIM (iSIM), can halve the resolution limit of conventional light microscopy.
However, unlike other super-resolution methods, iSIM can capture images at up to 100Hz, and with a greater degree of optical sectioning (rejection of out-of-focus light). This enables iSIM to generate 3D + time super-resolved images deep into biological structures, all while using conventional fluorescent dyes and molecules.


Super-Resolution Spinning Disk Confocal
Spinning disk confocal microscopy is a versatile and widely used imaging technique in biology due to its ability to perform fast, 3D imaging of live cells.
Recently, techniques have been created that combine super-resolution imaging with the simplicity and optical sectioning capability of spinning disk confocal, resulting in a spinning disk system capable of a twofold resolution improvement over the diffraction limit.
SOFI
Super-resolution optical fluctuation imaging (SOFI) offers high-speed and low-cost super-resolution imaging of samples through computational analysis of fluctuating fluorophores.
SOFI exploits the correlation of light from individual fluorophores, the more light and the more correlated the light that hits a pixel, the brighter that pixel will be in the SOFI image. By correlating fluorophores in time and/or space, SOFI is a powerful super-resolution technique.

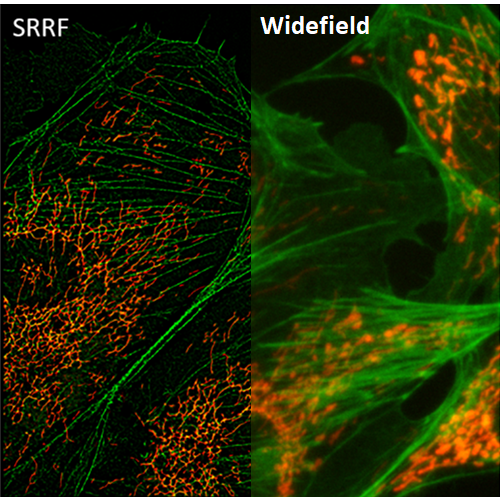
SRRF
Super-resolution radial fluctuations (SRRF) is a unique super-resolution algorithm that allows for fast live-cell imaging with standard fluorescent labels, when in samples with an ultra-high density of fluorophores.
SRRF processes images based on radial symmetry throughout image frames, due to circular point spread functions (PSFs). Spatial and temporal analysis allows SRRF to be a powerful platform for the super-resolution analysis of samples.
Expansion Microscopy
Rather than using optics or photophysics to separate biological structures from one another, expansion microscopy takes the opposite approach and physically enlarges the sample to pull structures further apart.
The result is a sample where biological structures are physically further apart than before. These structures can then be resolved even with conventional microscopy techniques.
Prime 95B sCMOS vs EMCCD For Super-Resolution Imaging
To support our claim that the Prime 95B is the best camera for super-resolution localization microscopy, we’ve collaborated with some of our customers to compare the localization accuracy of the Prime 95B to popular EMCCD cameras.
We also investigated the improvement made by PrimeEnhance™, the live denoising algorithm that accompanies the Prime 95B.
To perform these tests, DNA-origami nanostructures with a pre-defined pattern of fluorophores were provided by GATTAquant and used as precision standards.
