
Introduction
Fluorescence microscopy is a fundamental set of techniques in the life sciences for visualizing structures in living systems. Typically, a fluorescent molecule, either synthetic or biological, is associated with a structure of interest in a biological sample. When the sample is illuminated with light of an appropriate wavelength to excite the fluorophore, photons are emitted by the molecule which allows the molecule of interest to be visualized. The resolution at which fluorescent molecules can be visualized depends greatly on the preparation of the sample and the objectives used, but this can be limited by out of focus light being collected in a focal plane.
Total internal reflection fluorescence microscopy (TIRF) makes use of specific optics to produce illumination light only at the 50-100 nm range at the interface of the slide, massively reducing out of focus light and improving the ability to detect fluorescent molecules. Because of its low light intensity and high spatial resolution, it is a key technique in live-cell imaging.
Epifluorescence vs TIRF Microscopy
In traditional epifluorescence microscopy, illumination light is focused through the objective, exciting fluorescent molecules in the sample. The resultant emitted photons are collected by the same objective and focused onto a scientific camera for detection. This exposes the sample to excess and out of focus light which increases the effective light dose to the cells being imaged. Light itself, as well as radicals given off in the process of fluorescence, are toxic to cells and contribute to sample degradation. Further, fluorescence outside of the focal plane contributes to noise in the image, reducing overall signal to noise and spatial resolution. TIRF improves both of these elements through differences in the way illumination occurs (Figure 1).
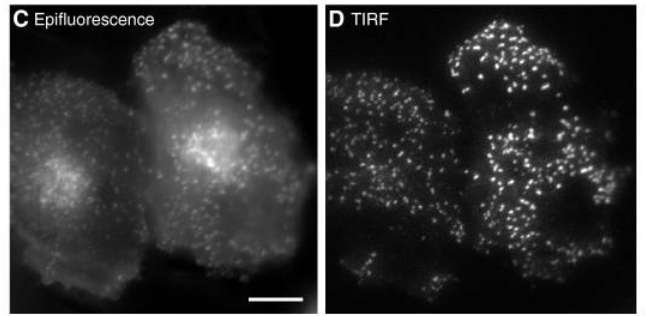
Left Epifluorescent illumination produces more out of focus light shown as diffuse signal in the cytoplasm.
Right TIRF illumination produces a higher signal to noise by producing an optical section only within the superficial 100 nm.
Scale bar: 10 μm. Adapted from Mattheyses et al. (2010).
In TIRF microscopy, the excitation light does not come from a focused objective as in epifluorescence microscopy, but from an obliquely angled source which may be through an objective or not. This gives an effective illumination of only the most superficial ~100 nm of the sample, greatly improving axial resolution (lateral resolution is still diffraction limited). The total light dose to the cell is minimized, reducing phototoxic effects, and removing out-of-focus fluorescence, gives a much higher signal-to-noise ratio.
Principles Of TIRF
When light traveling through a medium hits a medium with a different refractive index, some of the light will be reflected and some will be transmitted. The relative proportions depend on the differences in, amongst other things, the incident angle. If the light is traveling from a relatively high refractive index into a lower refractive index, there will be an angle beyond which all the light is reflected. This is the critical angle (ΘC) and is defined by Snell’s law:
Θ𝐶=sin−1(n1/n2 )
Where n1 and n2 are the refractive indices of the media respectively.
Equation 1: Snell’s law for the critical angle of light
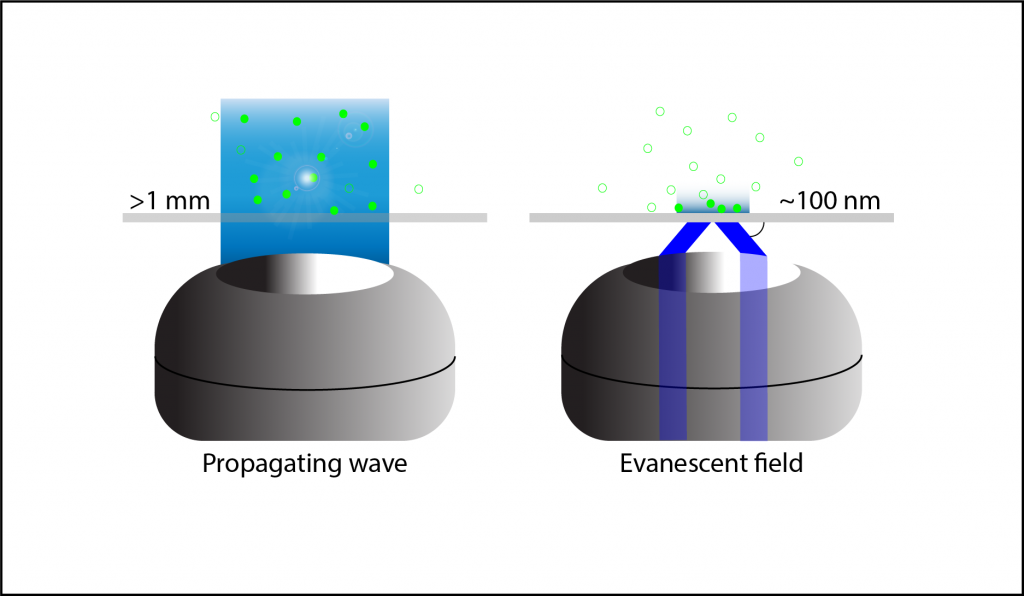
When light hits an interface beyond the critical angle it is completely reflected, this is called Total Internal Reflection (TIR). In TIRF microscopy, the light is presented to the slide-sample interface beyond this critical angle, either through the objective or using prisms. This produces an electromagnetic field at the interface called the evanescent field or wave (Figure 2).
The evanescent wave is the same wavelength as the light reflected during TIR but decays exponentially resulting in a very superficial excitation of fluorescence in the sample. Propagating waves, like the light used in epifluorescence microscopy, do not decay in this manner resulting in thicker volumes of excited samples. TIRF fluorescence can be detected, as with other fluorescent microscopy techniques, using the objective to focus the light and dichroic mirrors to separate the excitation and emission wavelengths.
TIRF Light Paths
The light path for TIRF can occur in many ways but the two main methods are: coupling the illumination and detection through the objective and separating illumination and detection using a prism (Figure 3).
Objective-based TIRF
Here, like in many fluorescent microscopes, the excitation and emission light go through a single objective. For TIRF, the light path through the objective must be precisely calibrated so the light exiting the optics hits the same beyond the critical angle.
This is achieved by making the laser light source entering the back-pupil of the objective off-axis. The angle of incidence leaving the lens correlates with the extent of off-axis input light. High numerical aperture lenses (>1.4 NA) are necessary to achieve sufficiently oblique angles from the lens to surpass the critical angle and produce Total Internal Reflection (TIR). The benefit of objective-based TIRF is that emission occurs at the interface, so no tissue or material distorts the emitted light back to the objective, and objective-based TIRF collects more light. Adjusting the angle of the illumination is harder than other methods, however, and there is less freedom to manipulate the evanescent field.
The TIRF Lens
Due to the specific nature of TIRF with objective illumination, specific lenses have been developed to facilitate TIRF microscopy. These have very high numerical apertures of 1.49-1.7 (for 60x and 100x respectively) which results in the highest incident angle and shallowest evanescent field. A common feature, which accounts for the increased temperature used in live cell imaging (37°C from 23°C), is a correction collar to compensate for the refractive index of immersion oil which changes over temperature. This prevents spherical aberrations which are induced by refractive index mismatching.
TIRF Through A Prism
Other methods of achieving TIRF are by uncoupling the light-source from the objective and instead producing sufficiently oblique light by using a set of prisms at the interface surface between the sample and the slide. The objective is usually on the opposite side of the sample to allow for the extra hardware needed. The benefit of this type of setup is the ease of adjusting the beam and angle of the light. A more oblique angle produces deeper penetration of the evanescent field and a thicker optical section. However, the thickness of the sample is a concern as it sits between the excitation light and the objective. This is best done with thin, transparent or cleared samples to avoid emission light scattering.
The benefits of prism-based TIRF systems are that the illumination light path can be directed so as not to enter the objective at all. The light path is more freely and easily manipulated for optimizing parameters like incident angle and laser alignment. A significant increase in signal-to-noise occurs as well. Emission filters to remove illumination light can be removed from the objective light path. The presence of a filter decreases signal as photons are always lost when passing through a lens, and illumination light contributes to noise signal.
In low-light applications of TIRF, such as single-molecule imaging, techniques have been developed which can combine the signal-to-noise benefits of prism-based systems, and the minimal light scattering of objective-based TIRF. Using wideband micromirrors, for example, between a fixed objective and the sample or after the objective back aperture to reflect illumination light out of the emission pathway. This can greatly increase the signal-to-noise.

TIRF Microscopy Applications
Live cell imaging can benefit greatly from the very thin optical section of the evanescent field. Conversely, this means that only investigation at or near the cell membrane can be achieved in cultured cells. For this reason, ligand-receptor binding, protein-membrane trafficking, synaptic vesicle fusion, and cell adhesion proteins have been extensively studied with TIRF microscopy in cultured cells as well as other events near the membrane such as actin dynamics in filopodia and G-protein coupled receptor transduction events. The reduction in luminance and low photo-toxicity allows for multiple exposures without affecting the health of the sample which is ideal for long time-lapse studies or fast, high temporal resolution imaging.
FRET
TIRF has been used in combination with Förster Resonance Energy Transfer (FRET) for dynamic imaging between fluorophores as they interact. This can be between different molecules within a complex, or in different domains of single molecules. FRET produces information about fluorophore proximity in the order of 2-10nm resolution. It is often interpreted as the change in relative fluorescence of, or between, the donor and acceptor. As such, it is very sensitive to noise and benefits greatly from the increased signal-to-noise ratio provided by TIRF. This is especially important for low light FRET applications such as single-molecule FRET.
TIRF And Super Resolution Microscopy (SIM, PALM, STORM)
In 2006, TIRF was successfully combined with Structured Illumination Microscopy (SIM) to further improve the lateral resolution of TIRF to super-resolution distances of ~90 nm (Figure 4). The addition of SIM, which uses phase and frequency information, provides depth information as well, enabling 3D imaging with high lateral and axial resolution.
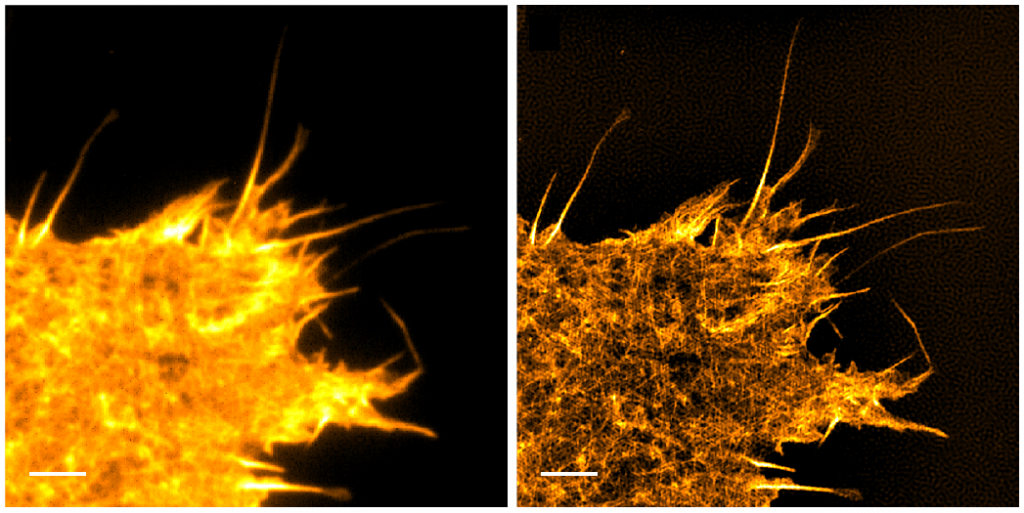
Left Expressing the fluorescent actin LifeAct GFP. Right SIM TIRF of the same field of view, providing an increase in lateral resolution and contrast by computing spatial information from phase and frequency data.
Scale bar: 3 μm. Adapted from Young et al. (2016).
Another key improvement was apochromatic multicolor imaging with TIRF. This was difficult for two reasons. Firstly, aligning multiple illumination paths to provide TIRF at the same spot was problematic. Secondly, the evanescent wave is dependent on the wavelength of the light and therefore different wavelengths at the same angle produce different relative amounts of excitation light, making interpretation difficult. This is now achieved with various spatial wave modulators and optoacoustic modulators to sculpt the light paths. Super-resolution TIRF, including SIM-TIRF using multiple light channels, is now routine.
TIRF has also been employed in combination with STochastic Optical Reconstruction Microscopy (STORM) and Photoactivated Localisation Microscopy (PALM), two techniques which rely on the computing the spatial distribution of single fluorophore molecules to deconvolve their position in space to a high precision. These techniques use TIRF to get a high signal to noise ratio in a very shallow optical section, improving localization accuracy.
Spinning-Spot TIRF
Further improvements of TIRF have come from manipulating TIRF itself, rather than through additional processing. Spinning-spot TIRF is an improvement over objective-based TIRF wherein the laser coming in at the periphery of the back focal plane of the lens is rotated around the circumference of the back focal plane in a donut shape (Figure 5). Instead of the illumination coming from a single angle, the beam is rotated faster than the exposure time of a frame, effectively averaging the illumination from all radial positions giving a uniform spot. The spinning illumination then averages out the inconsistencies in the field, producing clearer, higher signal-to-noise images than traditional TIRF. It, like regular TIRF, and be used in combination with other methods described above.
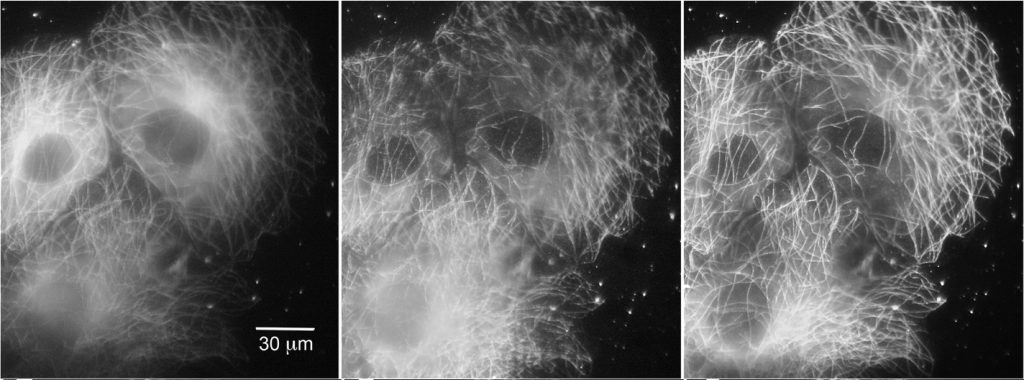
Left COS cells expressing tubulin tagged with EGFP, under epifluorescence, Middle Standard TIRF and Right Spinning spot TIRF illumination.
Scale bar: 30 μm. Adapted from Ellefsen et al. (2015).
Cameras For TIRF
The applications of TIRF microscopy are varied and include both low- and high-light applications. However, typically high spatial resolution and/or fast live imaging are motivators for using TIRF microscopy.
The technical necessity of using a high numerical aperture lens to achieve TIRF, coupled with the emphasis on high spatial resolution, means that TIRF users require a camera with appropriately sized pixels for Nyquist sampling at diffraction limited resolution whilst being sensitive enough for low-light single molecule fluorescence applications.
Sensitivity is often the first concern for TIRF imaging, with back-illuminated, 95% quantum efficient sCMOS and EMCCD sensors being the typical choice. To achieve diffraction limited resolution, the next concern is matching the pixel size to the magnification being used. A pixel size around 10-11 μm is optimized for a 100x lens and a pixel size around 6-7 μm is optimized for a 60x lens, both common pixel sizes of sCMOS cameras. EMCCD cameras typically have larger 13 or 16 μm pixels which would require 120x or 150x magnification respectively.
A large sensor area can also be important for some TIRF applications. sCMOS sensors are generally larger than EMCCD, where the most common EMCCDs have an 11 mm diagonal field of view and sCMOS sensors tend to range from 18 mm to 32 mm to better take advantage of modern, large field of view microscopes.
For high temporal resolution applications such as imaging protein dynamics or receptor trafficking, faster frame rates can be achieved with sCMOS cameras with less region cropping than EMCCDs to combine high speeds with large fields of view.
Summary
TIRF microscopy relies on the evanescent field generated by total internal reflection, to excite a very shallow <100 nm optical section at the interface between the surface and the sample. It has become a stand-out technique in live cell and single molecule imaging due to its exceptional axial resolution and low total luminance. In combination with other methods, such as FRET and super-resolution techniques, it is widely used to answer questions in the life sciences.
References
Deng, Y., & Asbury, C. L. (2017). Simultaneous Manipulation and Super-Resolution Fluorescence Imaging of Individual Kinetochores Coupled to Microtubule Tips. Methods in Molecular Biology (Clifton, N.J.), 1486, 437–467. http://doi.org/10.1007/978-1-4939-6421-5_17
Ellefsen, K. L., Dynes, J. L., & Parker, I. (2015). Spinning-Spot Shadowless TIRF Microscopy. PLoS ONE, 10(8), e0136055. http://doi.org/10.1371/journal.pone.0136055
Fish, K. N. (2009). Total Internal Reflection Fluorescence (TIRF) Microscopy. Current Protocols in Cytometry / Editorial Board, J. Paul Robinson, Managing Editor … [et Al.], 0 12, Unit12.18. http://doi.org/10.1002/0471142956.cy1218s50
Boulanger, J., Gueudry, C., Münch, D., Cinquin, B., Paul-Gilloteaux, P., Bardin, S., … Salamero, J. (2014). Fast high-resolution 3D total internal reflection fluorescence microscopy by incidence angle scanning and azimuthal averaging. Proceedings of the National Academy of Sciences of the United States of America, 111(48), 17164–17169. http://doi.org/10.1073/pnas.1414106111
Mattheyses, A. L., Simon, S. M. & Rappoport, J. Z. (2010) Imaging with total internal reflection fluorescence microscopy for the cell biologist. J Cell Sci. Nov 1; 123(21): 3621–3628. doi: 10.1242/jcs.056218
Raab, M., Jusuk, I., Molle, J., Buhr, E., Bodermann, B., Bergmann, D., … Tinnefeld, P. (2018). Using DNA origami nanorulers as traceable distance measurement standards and nanoscopic benchmark structures. Scientific Reports, 8, 1780. http://doi.org/10.1038/s41598-018-19905-x
Ramachandran, S., Cohen, D. A., Quist, A. P., & Lal, R. (2013). High performance, LED powered, waveguide based total internal reflection microscopy. Scientific Reports, 3, 2133. http://doi.org/10.1038/srep02133
Ross, S.T., Schwartz S., Fellers, T. J., Davidson, M. W., (2017) Total Internal Reflection Fluorescence (TIRF) Microscopy. Retrieved from https://www.microscopyu.com
Scott, B. L., Sochacki, K. A., Low-Nam, S. T., Bailey, E. M., Luu, Q., Hor, A., … Hoppe, A. D. (2018). Membrane bending occurs at all stages of clathrin-coat assembly and defines endocytic dynamics. Nature Communications, 9, 419. http://doi.org/10.1038/s41467-018-02818-8
Tabor, A., Möller, D., Hübner, H., Kornhuber, J., & Gmeiner, P. (2017). Visualization of ligand-induced dopamine D2S and D2L receptor internalization by TIRF microscopy. Scientific Reports, 7, 10894. http://doi.org/10.1038/s41598-017-11436-1
Thompson, N. L., Burghardt, T. P., & Axelrod, D. (1981). Measuring surface dynamics of biomolecules by total internal reflection fluorescence with photobleaching recovery or correlation spectroscopy. Biophysical Journal, 33(3), 435–454.
Weigel, A. V., Tamkun, M. M., & Krapf, D. (2013). Quantifying the dynamic interactions between a clathrin-coated pit and cargo molecules. Proceedings of the National Academy of Sciences of the United States of America, 110(48), E4591–E4600. http://doi.org/10.1073/pnas.1315202110
Young, L. J., Ströhl, F., & Kaminski, C. F. (2016). A Guide to Structured Illumination TIRF Microscopy at High Speed with Multiple Colors. Journal of Visualized Experiments : JoVE, (111), 53988. Advance online publication. http://doi.org/10.3791/53988