
Introduction
A splitter is an optical device that splits light (such as laser beams) into two (or more) beams. Splitters are very useful in microscopy as they can act as an interface between the microscope and the detector/camera, splitting the emission light from the microscope.
There are two major types of splitters, emission image splitters that split light across a single camera sensor so that multiple wavelengths can be imaged simultaneously by one camera, and multiple camera adapters that split light to multiple cameras, allowing one sample to be imaged simultaneously by multiple cameras. Both these splitter devices essentially allow for simultaneous imaging of a sample at multiple wavelengths, polarization states or amplitudes. For an introduction to splitters, please refer to our technical note: “Introduction to splitters”.
This article will describe how such splitter devices can be used to enhance microscopy techniques and advanced imaging applications. Why use splitters?
Techniques Requiring Splitters
Splitters allow for simultaneous imaging of different states: rather than having to adjust microscope settings manually or with electronic switches, two states are optically available with no delay. An example of how this can be useful for imaging applications is multifocal plane microscopy (MPM), as seen in Fig.1. MPM involves imaging two different focal planes of the same sample simultaneously, as opposed to adjusting focus manually and then taking images at different desired focal planes.
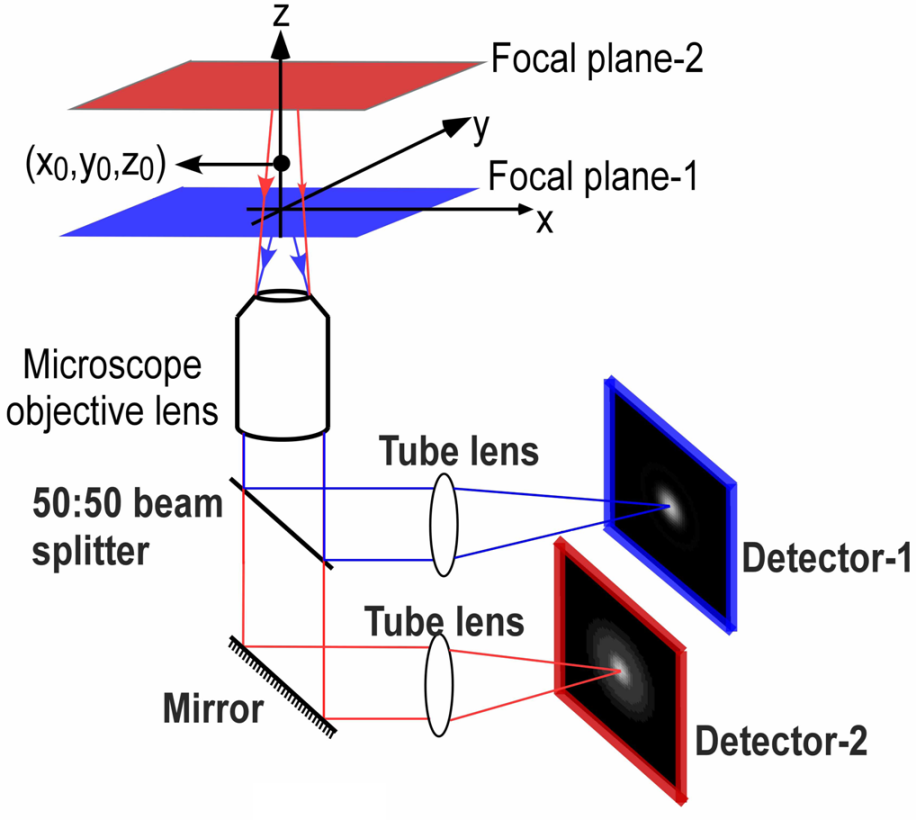
If imaging a 2D cell culture, one focal plane is usually sufficient as most cell preparations are not very thick. But as more researchers move to image larger 3D samples, MPM presents a useful way to image multiple desired focal planes as cells interact in a 3D scaffold, or as larger organisms make dynamic movements. High speeds are necessary when imaging live 3D samples, and by not having to manually adjust the focus, MPM presents a high-speed option for imaging multiple z-planes through a sample simultaneously. An example of MPM in action can be seen in Fig.2.

Without a splitter, time may be wasted while waiting for the sample to be moved to the correct position, such as the scanning seen in light-sheet techniques like selective plane illumination microscopy (SPIM). Scanning can also introduce smear-like effects due to the camera shutter and restrict the available exposure times. By capturing multiple points of interest simultaneously, these restrictions are avoided, allowing for the capture of small, faint and fast-moving subjects in a 3D space. MPM would not be possible without a splitter and demonstrates the power and flexibility of manipulating the emission light from a sample.
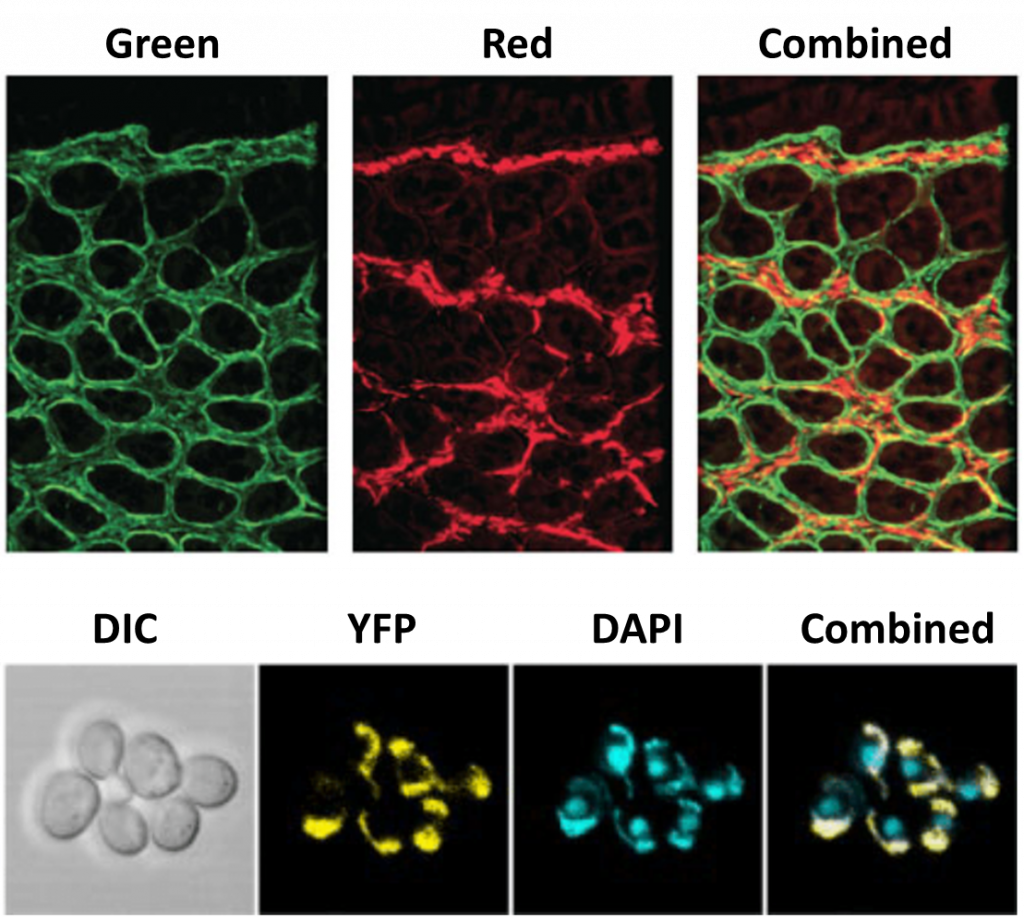
Another technique requiring a splitter is simultaneous multi-probe fluorescent imaging, involving simultaneous imaging of different fluorescent probes. The vast majority of fluorescence microscopy uses multiple fluorophores, especially on cells where the blue channel is usually occupied by DAPI (an easy-to-use marker for the cell nucleus), leaving a red and green channel for other fluorophores marking specific proteins.
Microscopes usually have an electronic switch so that these different fluorophores can be imaged in different channels, resulting in multi-channel images. There is still a time delay while switching, and with a splitter there is the option to image multiple fluorescent wavelengths simultaneously, eliminating any delay. This is especially useful when performing fluorescent imaging on live cells, where fast dynamic events can be captured at high speed. As well as combining fluorescent probes, brightfield images such as differential interference contrast (DIC) can also be combined with fluorescence images.
Techniques Enhanced By Splitters
Some techniques do not inherently need a splitter to function but greatly benefit from its inclusion. This is mostly due to the time-saving ability to image multiple different states simultaneously, with other advantages differing depending on the technique. It is important to note that virtually every advanced microscopy application benefits from a splitter and the ability to image more flexibly, so the techniques mentioned here are just the tip of the iceberg and this article is by no means exhaustive.
One such technique is Förster resonance energy transfer (FRET), a technique used to determine whether two fluorophores are within a certain distance of each other. FRET takes advantage of the principle of energy transfer between fluorophore molecules: similar to how two magnets will only attract once they are close enough together, two fluorophores will only exchange energy once they are within 1-10 nm of each other. In FRET there are two fluorescent molecules, a donor and an acceptor, if the donor is excited and moves close enough to the acceptor, it will transfer energy. The subsequent decrease in donor fluorescence and increase in acceptor fluorescence can be easily detected by a microscope. This essentially makes FRET a ‘spectroscopic ruler’ that can measure the distance between molecules, as the reaction only occurs if the fluorophores are within 10 nm of each other.
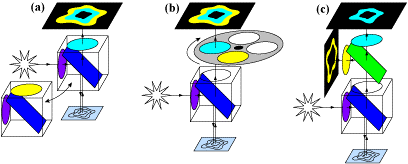
As there are two fluorophores, the light used to excite them should be at different wavelengths, otherwise, both would be activated at once and FRET would not occur. This means two specific wavelengths are needed to treat both fluorophores, and being able to image two different wavelengths would make this technique faster and more efficient, as it would be instantly clear when one decreases and the other increases in fluorescence intensity. FRET is an excellent technique to make use of a splitter, seen in Fig.4.
While all three methods in Fig.4 make use of splitters at some point, by using a multiple-camera adapter (Fig.4C) both fluorophores can be images simultaneously, making this version the fastest and most suitable for live-cell imaging. With no moving parts and no artifacts, simultaneous FRET with a splitter device enhances the technique and allows for better experimentation, improving imaging for dynamic processes.
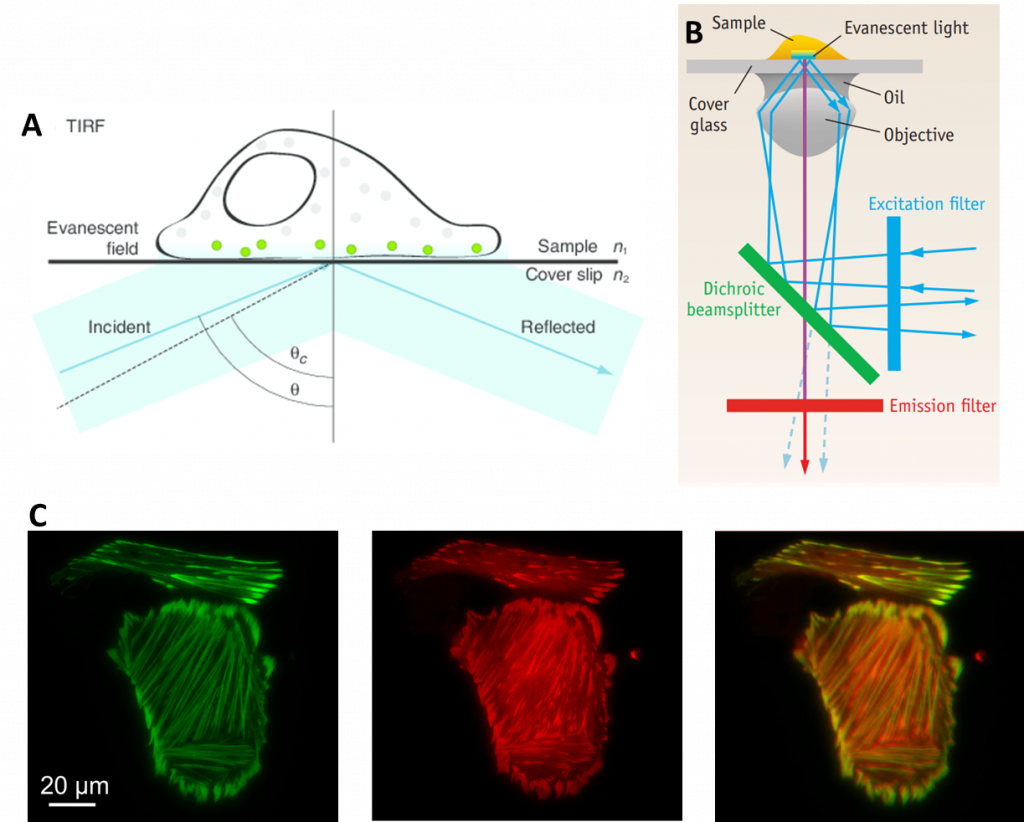
Another technique that benefits from splitters are total internal reflection fluorescence (TIRF), which involves reflecting light off a surface to only image a small portion of a sample. In TIRF, the excitation laser is angled in such a way that it completely reflects off of the glass sample slide. The result is that all excitation light is reflected away from the sample. However, in the area where the light is reflected, a small extension of the light spreads into a tiny section of the sample. This extension of light is known as an evanescent wave, and it typically only penetrates around 100 nm into the sample, depending on the microscope optics. The evanescent wave has the same wavelength as the reflected light so it still excites any fluorescent molecules within this small area.
Thanks to the small section of the sample that is illuminated, there is far less out-of-focus light and a better signal to noise ratio when compared to standard widefield fluorescence. As TIRF is restricted to a single focal plane (as only ~100 nm of sample is illuminated), it is particularly useful for studying areas of the sample close to the surface such as molecules immobilized on a coverslip or on the surface of cell membranes.
TIRF is routinely used for imaging dynamic processes, due to the short exposure times and single focal plane. In order to make this technique even faster and able to image multiple fluorophores, splitters have been used to great success, making TIRF more powerful. With a splitter, multi-channel TIRF can be performed simultaneously (as seen in Fig.5C), optimizing the technique for very rapid events.
Summary
The use of splitters both allows for new techniques to emerge while enhancing existing techniques, making splitter devices a powerful interface between microscope and camera, elevating researchers’ ability to control light and design more flexible and elegant experiments.